16S rRNA Microbiome Analysis - Part 1
Overview
Pipeline Outline
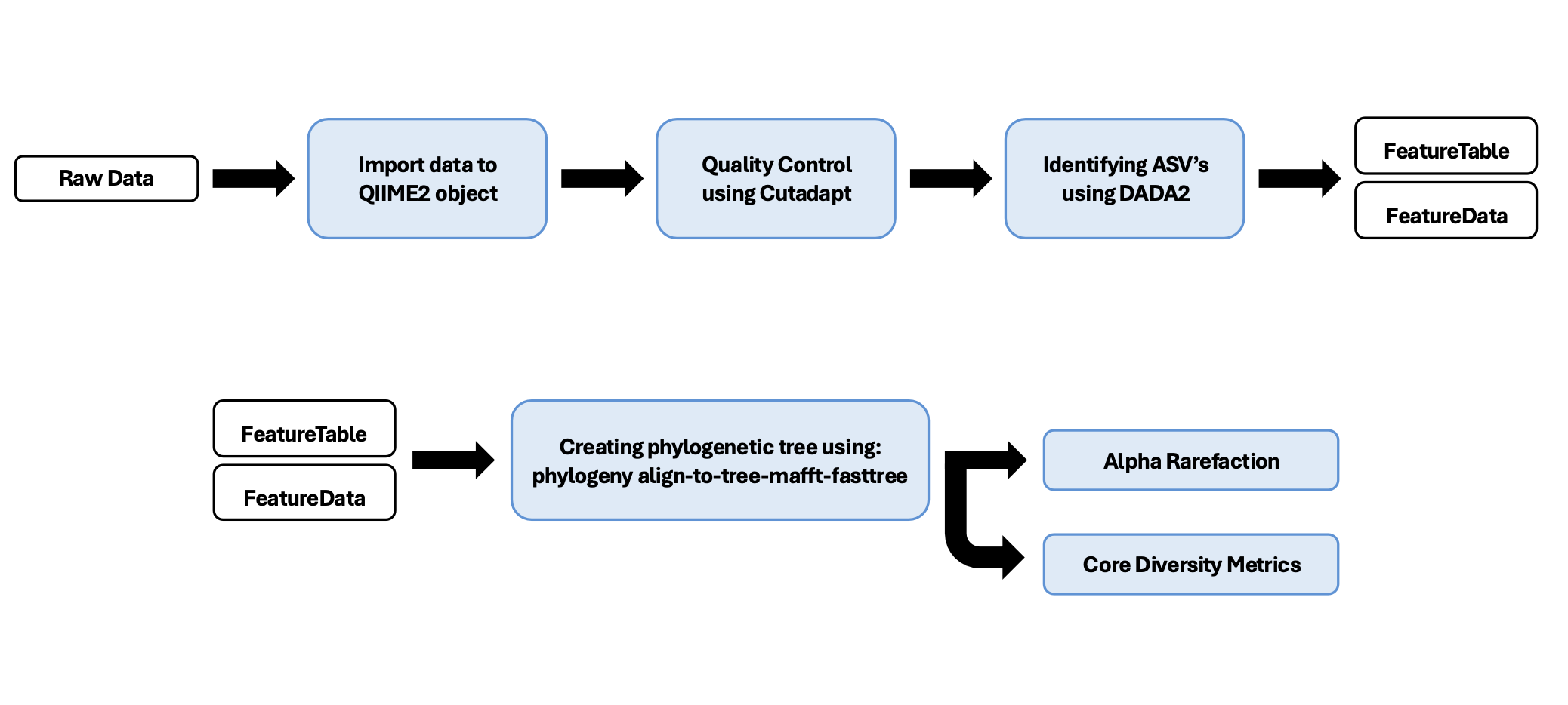
0-File Management and Making Manifest for Importing
qiime_out_dir="output"
mkdir $qiime_out_dirqiime metadata tabulate \
--m-input-file sample-metadata.tsv \
--o-visualization sample-metadata-viz.qzv1-Importing Sequencing Data to QIIME Object
import_seqs_dir="output/1_imported_seqs"
manifest_file= ""
mkdir $import_seqs_dir
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path $manifest_file \
--input-format PairedEndFastqManifestPhred33V2 \
--output-path $import_seqs_dir/demux-pairedend.qza
qiime demux summarize \
--i-data $import_seqs_dir/demux-pairedend.qza \
--o-visualization $import_seqs_dir/demux-pairedend.qzv2-Quality Control of Sequences
input_seqs="output/1_imported_seqs/demux-pairedend.qza"
f_primer=""
r_primer=""
error_rate=0.1
min_length=100
num_cores=22
trimmed_dir="output/2_trimmed_seqs"
mkdir $trimmed_dir
qiime cutadapt trim-paired \
--i-demultiplexed-sequences $input_seqs \
--p-cores $num_cores \
--p-front-f $f_primer \
--p-front-r $r_primer \
--p-error-rate $error_rate \
--p-minimum-length $min_length \
--o-trimmed-sequences $trimmed_dir/trimmed-demux-pairedend.qza \
--verbose \
&> $trimmed_dir/trimmed.log
qiime demux summarize \
--i-data $trimmed_dir/trimmed-demux-pairedend.qza \
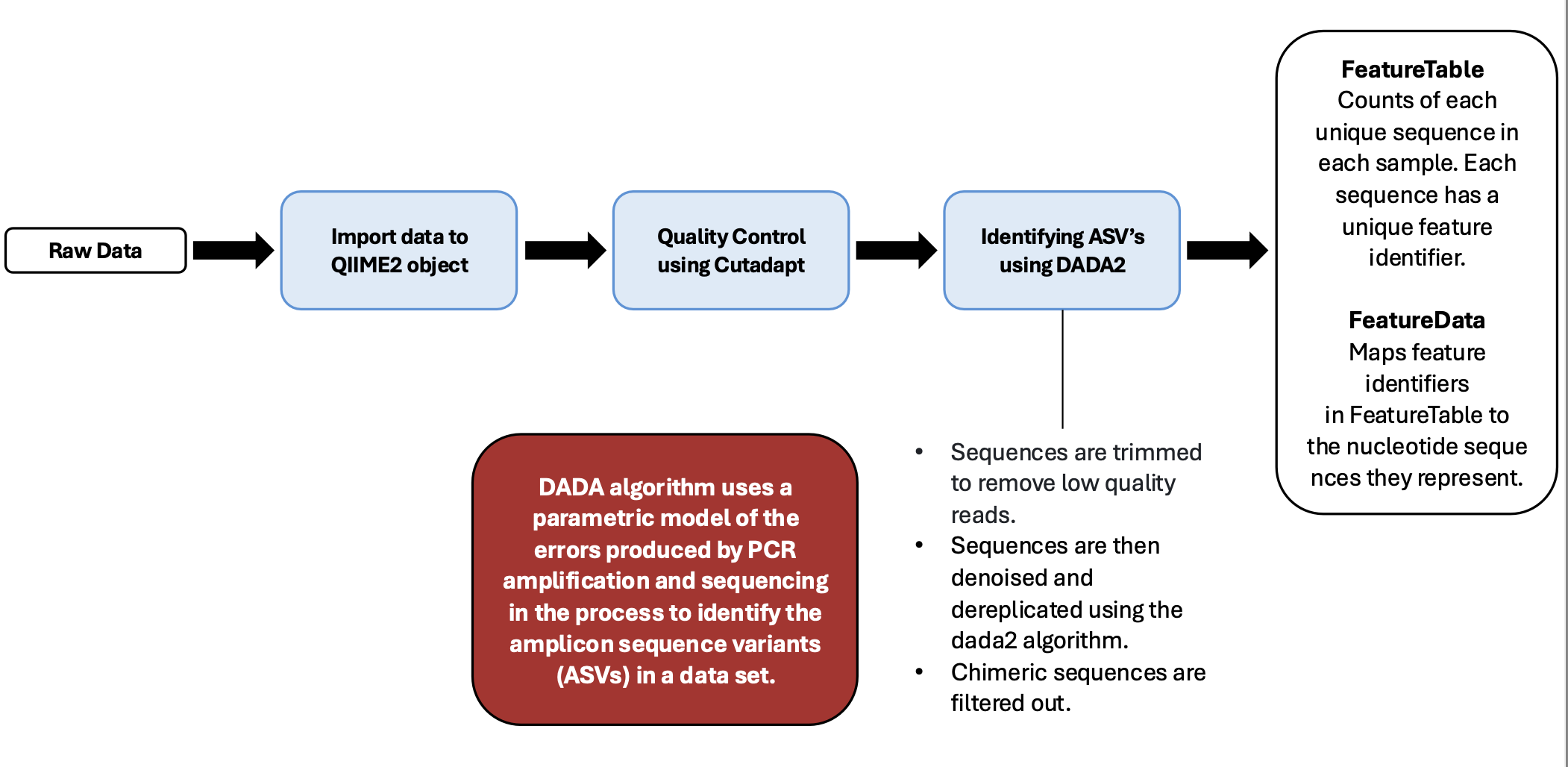
--o-visualization $trimmed_dir/trimmed-demux-pairedend.qzv 3-Feature Table Construction of Amplicon Variance Sequences (ASVs) using DADA2
input_trimmed_seqs="output/2_trimmed_seqs/trimmed-demux-pairedend.qza"
num_cores=20
num_reads_learn=1000000
trunc_len_f=282
trunc_len_r=234
dada2_res_dir="output/3_denoised_seqs"
mkdir $dada2_res_dir
qiime dada2 denoise-paired \
--i-demultiplexed-seqs $input_trimmed_seqs \
--p-trunc-len-f $trunc_len_f \
--p-trunc-len-r $trunc_len_r \
--p-n-threads $num_cores \
--p-n-reads-learn $num_reads_learn \
--o-table $dada2_res_dir/feat-table.qza \
--o-representative-sequences $dada2_res_dir/rep-seqs.qza \
--o-denoising-stats $dada2_res_dir/denoising-stats.qza \
--verbose \
&> $dada2_res_dir/denoising_dada2.log
qiime metadata tabulate \
--m-input-file $dada2_res_dir/denoising-stats.qza \
--o-visualization $dada2_res_dir/denoising-stats.qzv
qiime feature-table summarize \
--i-table $dada2_res_dir/feat-table.qza \
--o-visualization $dada2_res_dir/feat-table.qzv
qiime feature-table tabulate-seqs \
--i-data $dada2_res_dir/rep-seqs.qza \
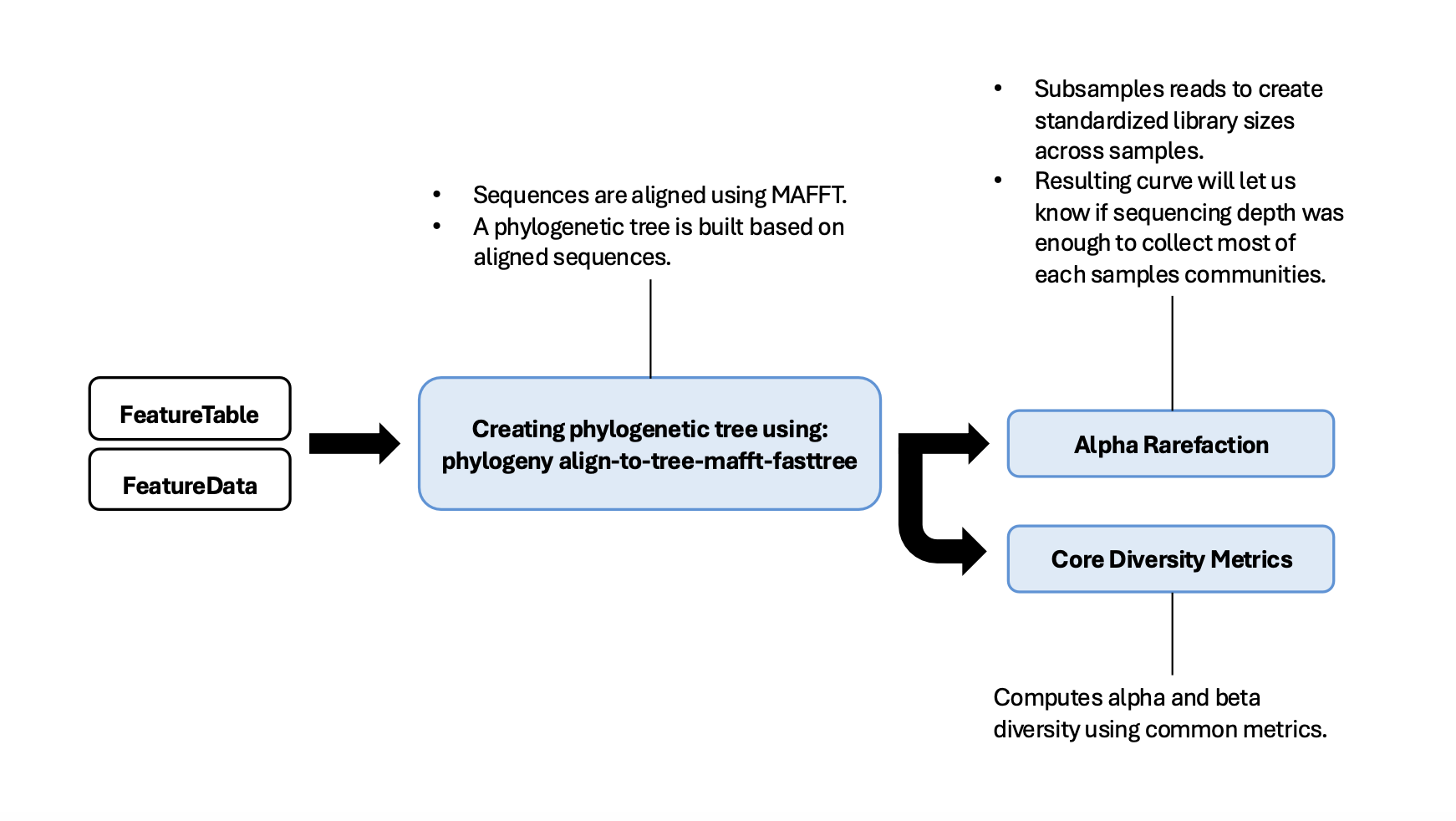
--o-visualization $dada2_res_dir/rep-seqs.qzv4-Phylogenectic Tree
phylo_tree_dir="output/4_phylogenetic_tree"
mkdir $phylo_tree_dir
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences $dada2_res_dir/rep-seqs.qza \
--o-alignment $phylo_tree_dir/aligned-rep-seqs.qza \
--o-masked-alignment $phylo_tree_dir/masked-aligned-rep-seqs.qza \
--o-tree $phylo_tree_dir/unrooted-tree.qza \
--o-rooted-tree $phylo_tree_dir/rooted-tree.qza5-Alpha Diversity, Beta Diversity, and Alpha Refraction
alpha_beta_dir="output/5_alpha_beta_div_rarefac"
mkdir $alpha_beta_dir
qiime diversity core-metrics-phylogenetic \
--i-phylogeny $phylo_tree_dir/rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 1103 \
--m-metadata-file sample-metadata.tsv \
--output-dir $alpha_beta_dir/diversity-core-metrics-phylogeneticqiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 4000 \
--m-metadata-file sample-metadata.tsv \
--o-visualization alpha-rarefaction.qzvFULL SCRIPT
#!/bin/bash
#SBATCH --partition=ircfhp
#SBATCH --nodelist=c923
#SBATCH --container=el9hw
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=10
#SBATCH --mem=30G
#SBATCH --chdir=/home
#SBATCH --output=qc_%A_%a_stdout.txt
#SBATCH --error=qc_%A_%a_stderr.txt
#SBATCH --mail-user=eleana-cabello@ouhsc.edu
#SBATCH --mail-type=ALL
#==============================================================================
# BASH Strict mode (i.e. "fail fast" to reduce hard-to-find bugs)
set -e # EXIT the script if any command returns non-zero exit status.
set -E # Make ERR trapping work inside functions too.
set -u # Variables must be pre-defined before using them.
set -o pipefail # If a pipe fails, returns the error code for the failed pipe
# even if it isn't the last command in a series of pipes.
#==============================================================================
module load QIIME2/2021.8
################################################
STEP 0
################################################
qiime_out_dir="output"
mkdir $qiime_out_dir
qiime metadata tabulate \
--m-input-file sample-metadata.tsv \
--o-visualization sample-metadata-viz.qzv
################################################
STEP 1
################################################
import_seqs_dir="output/1_imported_seqs"
manifest_file= ""
mkdir $import_seqs_dir
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path $manifest_file \
--input-format PairedEndFastqManifestPhred33V2 \
--output-path $import_seqs_dir/demux-pairedend.qza
qiime demux summarize \
--i-data $import_seqs_dir/demux-pairedend.qza \
--o-visualization $import_seqs_dir/demux-pairedend.qzv
################################################
STEP 2
################################################
input_seqs="output/1_imported_seqs/demux-pairedend.qza"
f_primer=""
r_primer=""
error_rate=0.1
min_length=100
num_cores=22
trimmed_dir="output/2_trimmed_seqs"
mkdir $trimmed_dir
qiime cutadapt trim-paired \
--i-demultiplexed-sequences $input_seqs \
--p-cores $num_cores \
--p-front-f $f_primer \
--p-front-r $r_primer \
--p-error-rate $error_rate \
--p-minimum-length $min_length \
--o-trimmed-sequences $trimmed_dir/trimmed-demux-pairedend.qza \
--verbose \
&> $trimmed_dir/trimmed.log
qiime demux summarize \
--i-data $trimmed_dir/trimmed-demux-pairedend.qza \
--o-visualization $trimmed_dir/trimmed-demux-pairedend.qzv
################################################
STEP 3
################################################
input_trimmed_seqs="output/2_trimmed_seqs/trimmed-demux-pairedend.qza"
num_cores=20
num_reads_learn=1000000
trunc_len_f=282
trunc_len_r=234
dada2_res_dir="output/3_denoised_seqs"
mkdir $dada2_res_dir
qiime dada2 denoise-paired \
--i-demultiplexed-seqs $input_trimmed_seqs \
--p-trunc-len-f $trunc_len_f \
--p-trunc-len-r $trunc_len_r \
--p-n-threads $num_cores \
--p-n-reads-learn $num_reads_learn \
--o-table $dada2_res_dir/feat-table.qza \
--o-representative-sequences $dada2_res_dir/rep-seqs.qza \
--o-denoising-stats $dada2_res_dir/denoising-stats.qza \
--verbose \
&> $dada2_res_dir/denoising_dada2.log
qiime metadata tabulate \
--m-input-file $dada2_res_dir/denoising-stats.qza \
--o-visualization $dada2_res_dir/denoising-stats.qzv
qiime feature-table summarize \
--i-table $dada2_res_dir/feat-table.qza \
--o-visualization $dada2_res_dir/feat-table.qzv
qiime feature-table tabulate-seqs \
--i-data $dada2_res_dir/rep-seqs.qza \
--o-visualization $dada2_res_dir/rep-seqs.qzv
################################################
STEP 4
################################################
phylo_tree_dir="output/4_phylogenetic_tree"
mkdir $phylo_tree_dir
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences $dada2_res_dir/rep-seqs.qza \
--o-alignment $phylo_tree_dir/aligned-rep-seqs.qza \
--o-masked-alignment $phylo_tree_dir/masked-aligned-rep-seqs.qza \
--o-tree $phylo_tree_dir/unrooted-tree.qza \
--o-rooted-tree $phylo_tree_dir/rooted-tree.qza
################################################
STEP 5
################################################
alpha_beta_dir="output/5_alpha_beta_div_rarefac"
mkdir $alpha_beta_dir
sampling_depth=""
qiime diversity core-metrics-phylogenetic \
--i-phylogeny $phylo_tree_dir/rooted-tree.qza \
--i-table $dada2_res_dir/feat-table.qza \
--p-sampling-depth $sampling_depth \
--m-metadata-file ${manifest_file} \
--output-dir $alpha_beta_dir/diversity-core-metrics-phylogenetic
max_depth=""
qiime diversity alpha-rarefaction \
--i-table $dada2_res_dir/feat-table.qza \
--i-phylogeny $phylo_tree_dir/rooted-tree.qza \
--p-max-depth $max_depth \
--m-metadata-file ${manifest_file} \
--o-visualization $alpha_beta_dir/alpha-rarefaction.qzv