Quality Check of FASTQ Files on OSCER
FASTQ Files
The FASTQ file format is the defacto format for unmapped sequence reads generated from next-generation sequencing technologies.
The image belows shows the structure for one read entry in a FASTQ file, its important to note these files will have a million reads meaning millions of entries repeating this same structure:

Each entry can be broken down to four lines. Line 1 will always start with the @ symbol followed by sequence identifiers like instrument ID, run number, flow cell ID, etc. Line 2 will be the sequence itself. Line 3 will always start with the + symbol and on occasion be followed by the same information on line 1. It acts as a seperator between lines 2 and 4. Line 4 will contain the quality score for each nucleotide in the sequnce of Line 2, encoded in ASCII characters.

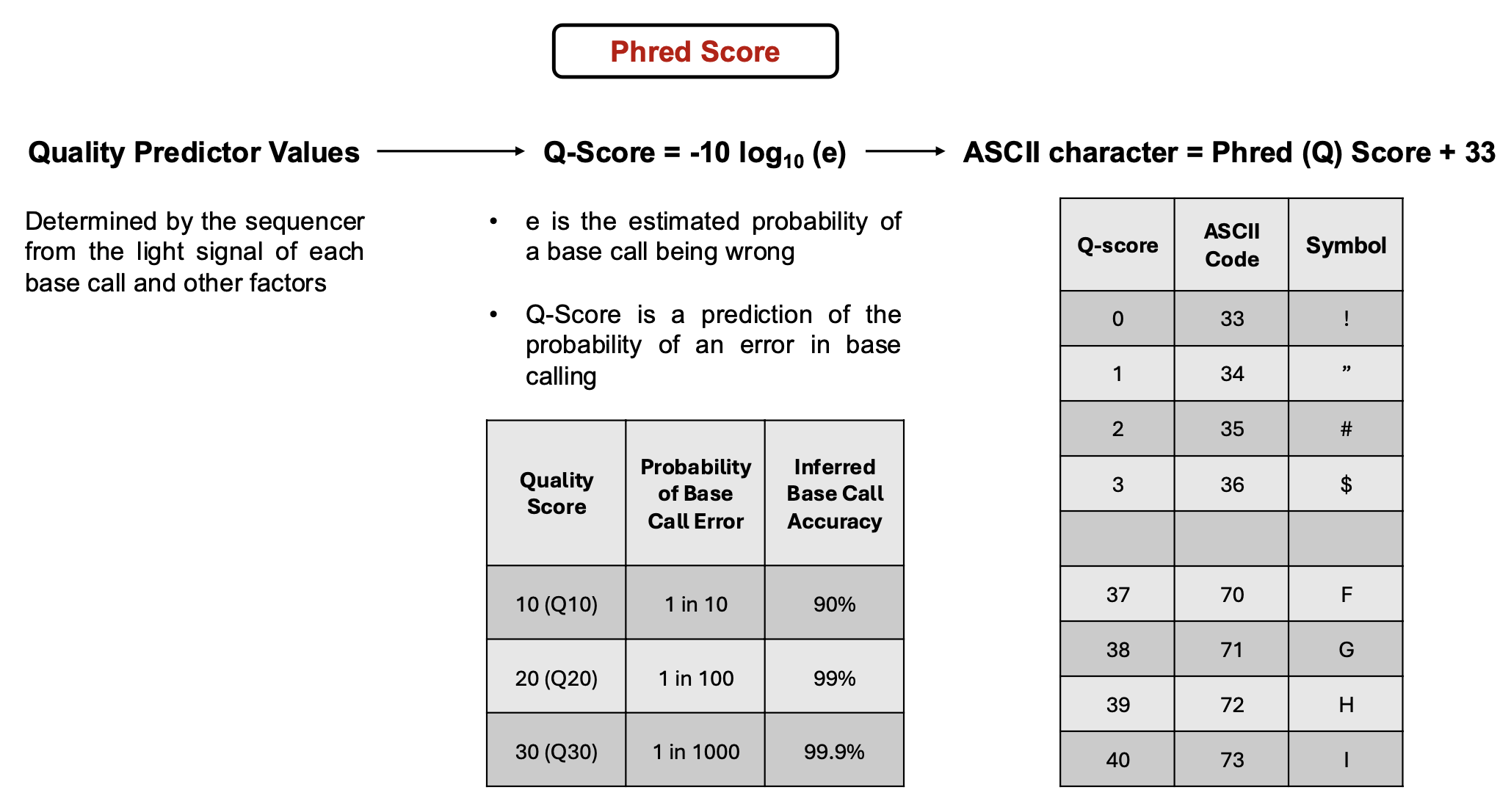
While reads are getting sequenced in a sequencer, quality predictor values are being created for each base call determined by the light signal emitted of each base, sign-to-noise ratio, and other factors determined by the machine. These Values are then used to calculate a Phred (Q) Score that takes into account the probability of a base call being incorrect and gives a prediction on probability of an error occuring in the base calling by the machine. The higher the Q-Score the less likely a base call is incorrect. To simplify this information to add to FASTQ files, an ASCII character is used and found by adding 33 to the Q-Score. The summed number and the ASCII chart is then used to find a character to allocate to the quality of each base.
Programs to Perform Quality Checks of FASTQs
FastQC
FastQC is a program that allows you to run a quick quality check on a fastq file (.fastq or .fastq.gz). To run this program store all fastq files in one directory. Then pass that directory to the FastQC command when running it through the terminal.
Let’s say you stored all your files in a directory called raw_data and you want to store results in a new directory called fastqc_results, you’re command would look like:
fastqc -o fastqc_results raw_data/*.fastq.gzEach fastq file processed will have a resulting _fastqc.html and _fastqc.zip. Below are examples of the results stored in a _fastqc.html file:
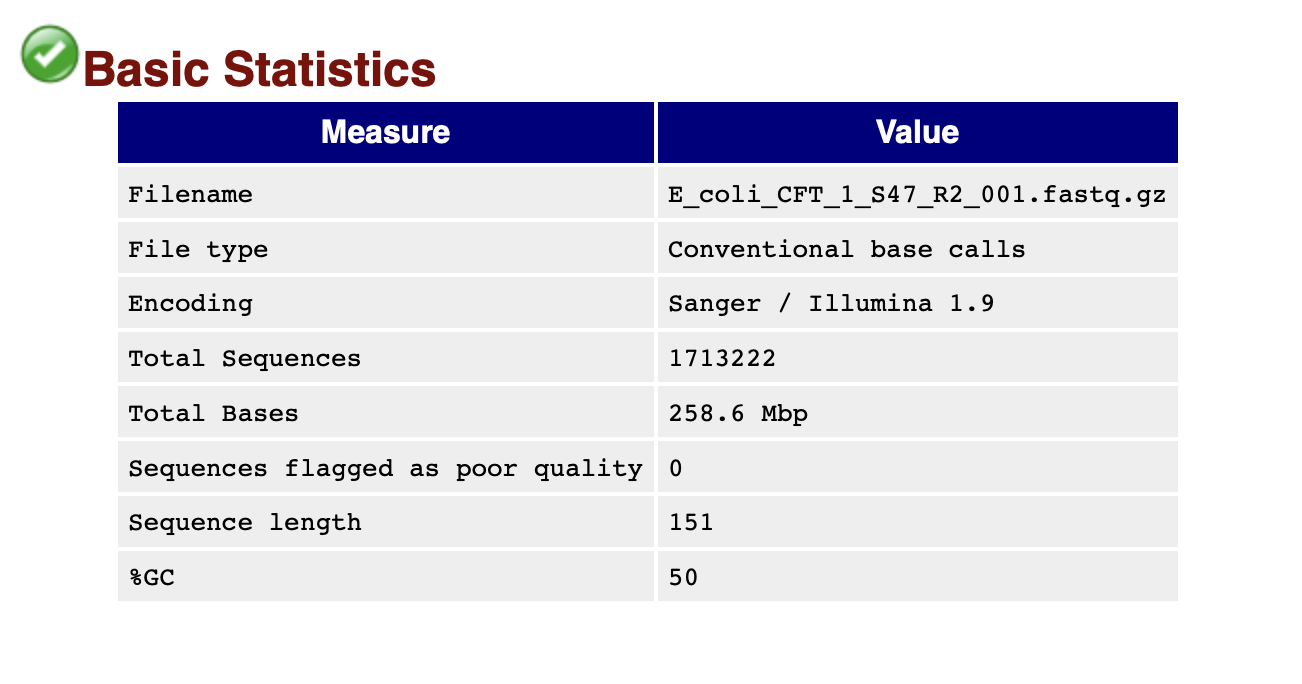
The basic statistics section provides a quick description of the file including name, total number of sequences, total bases, range of sequence lengths, and GC percentage.
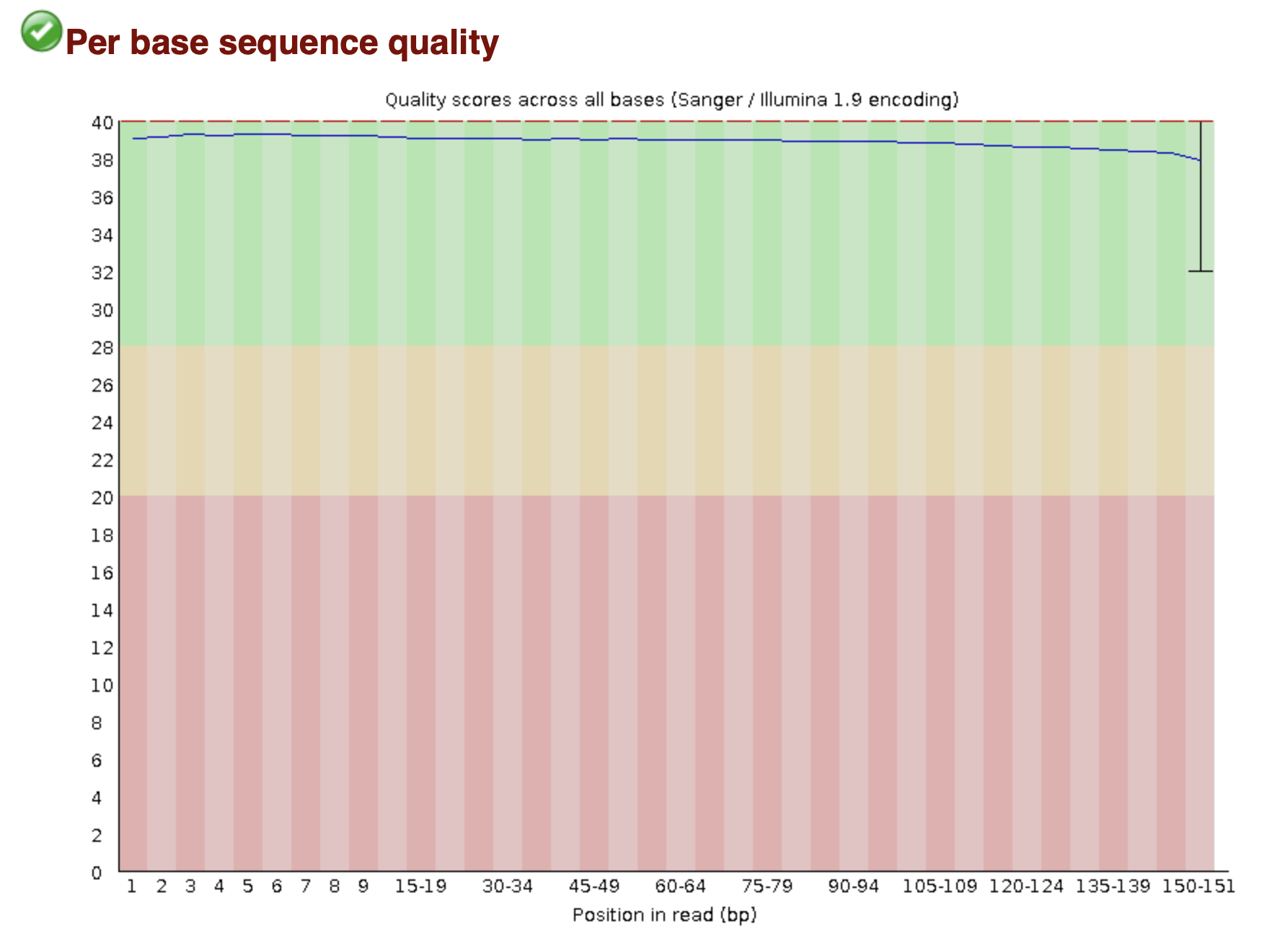
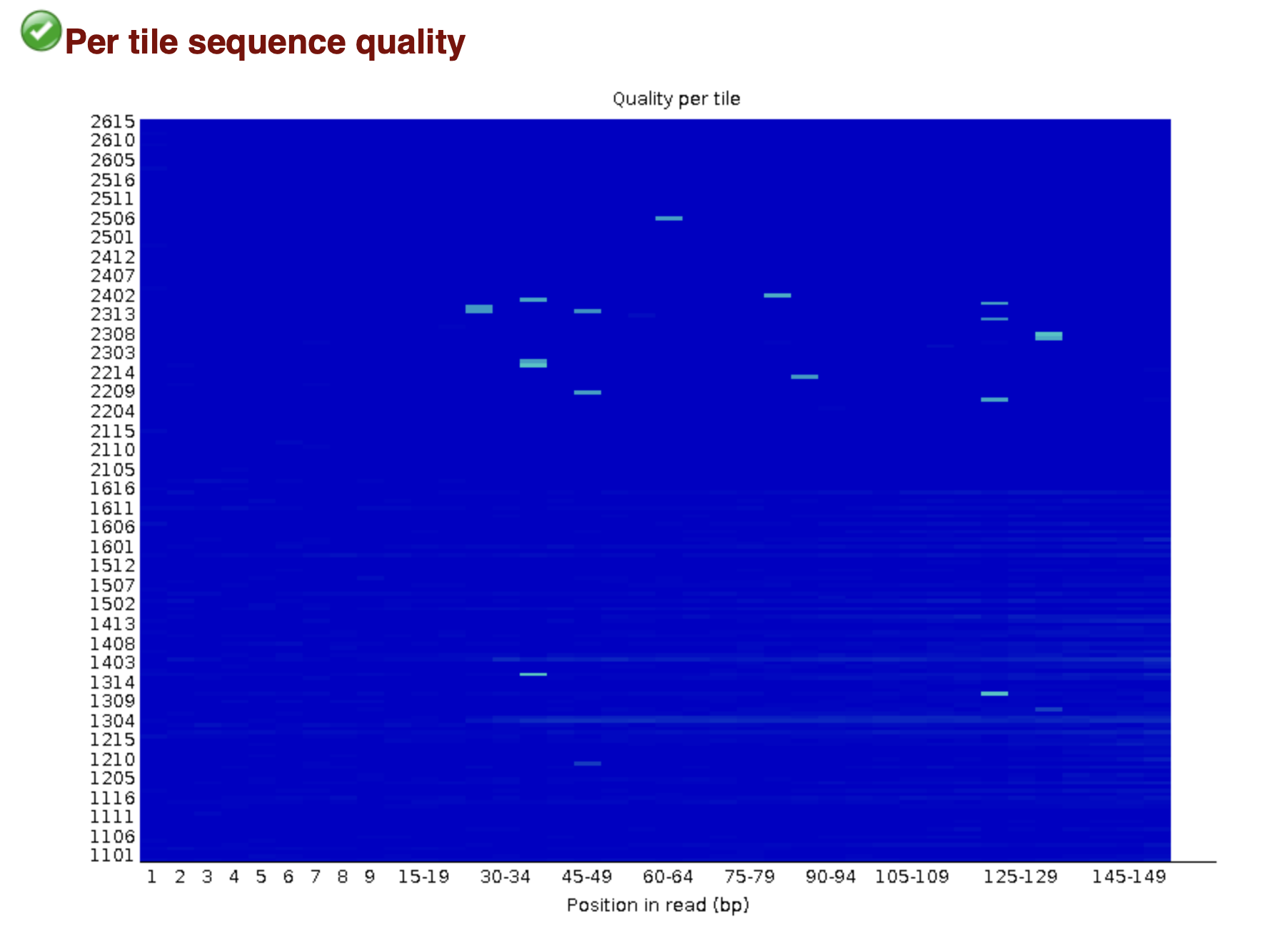
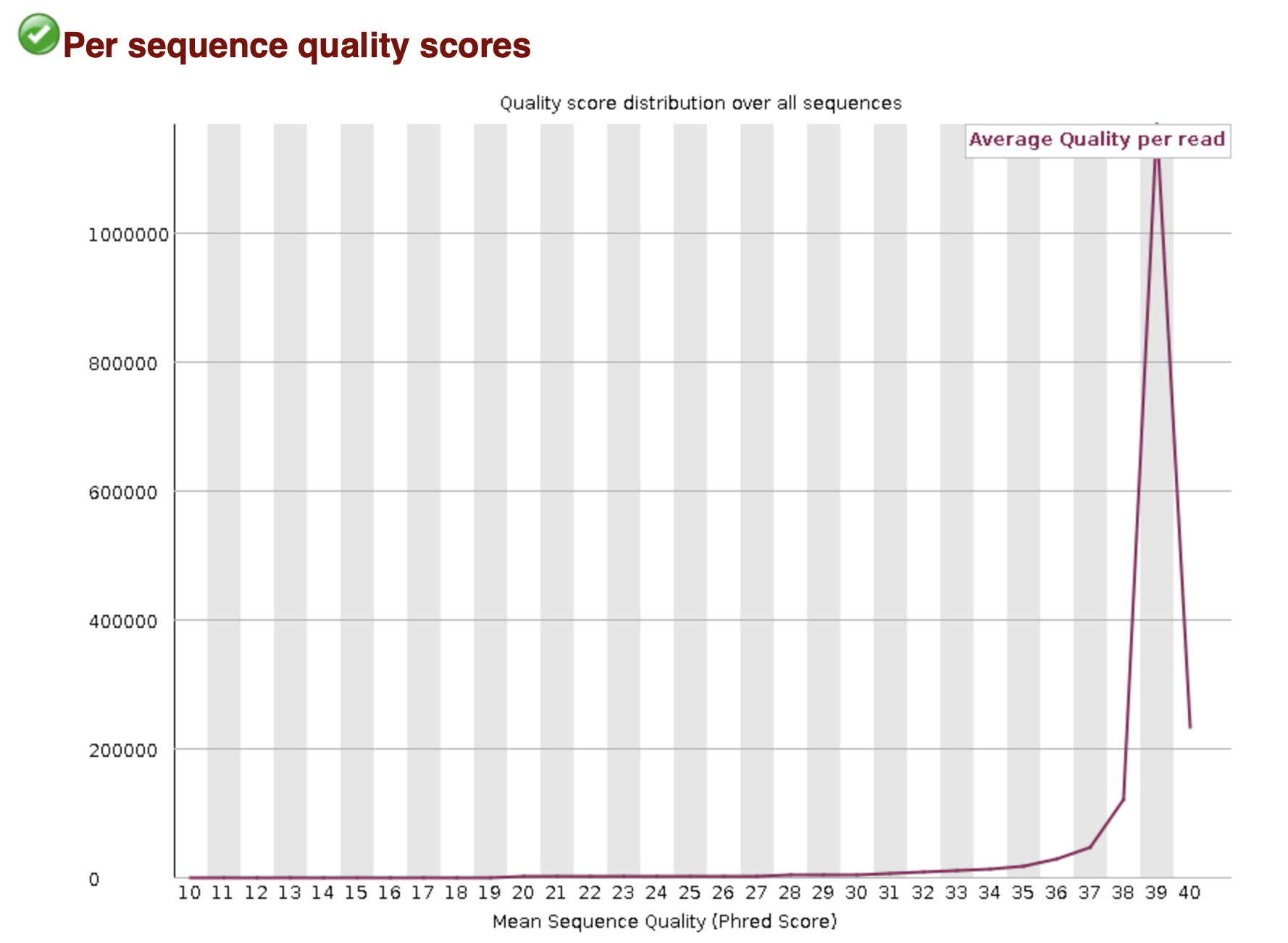
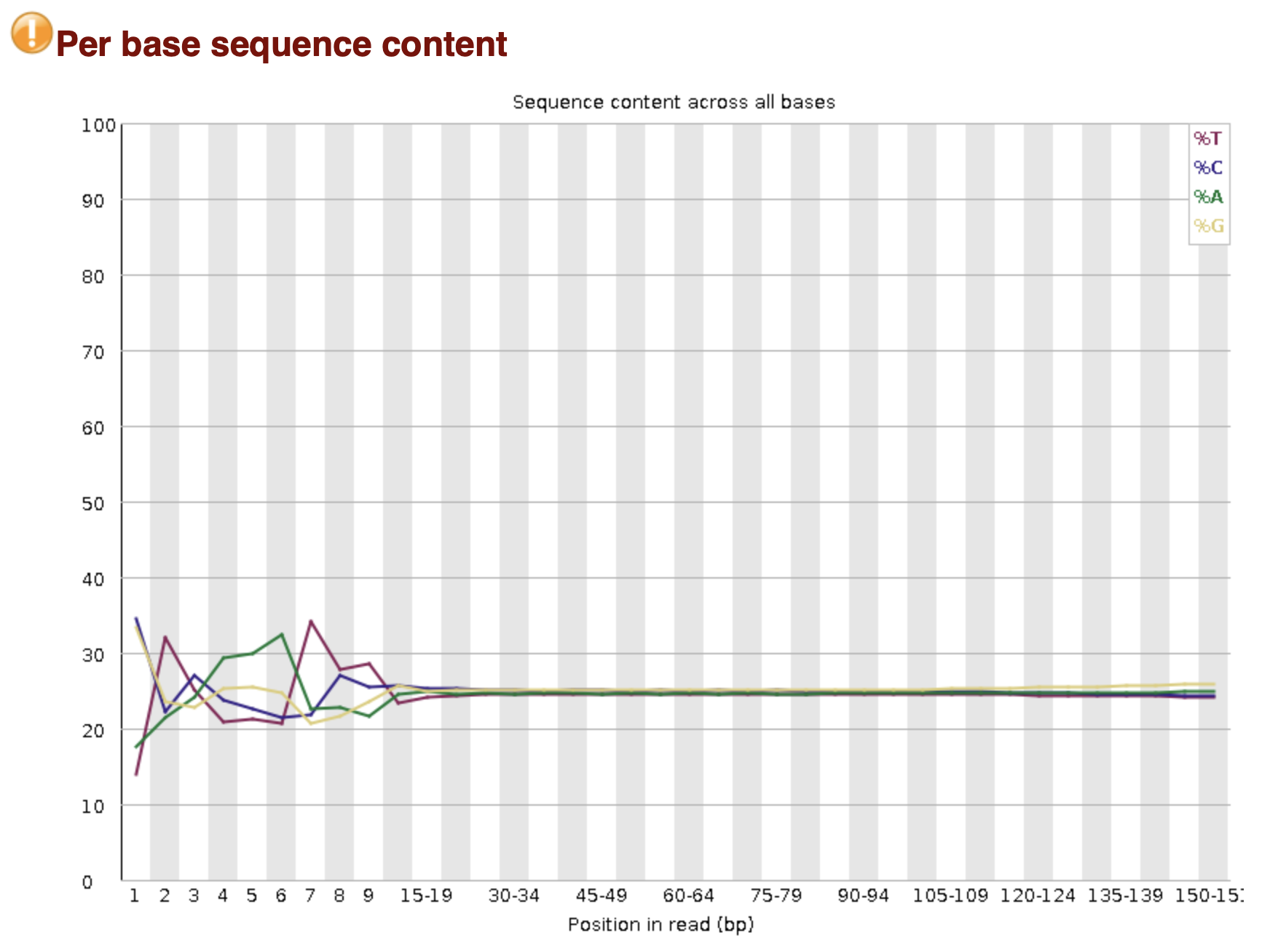
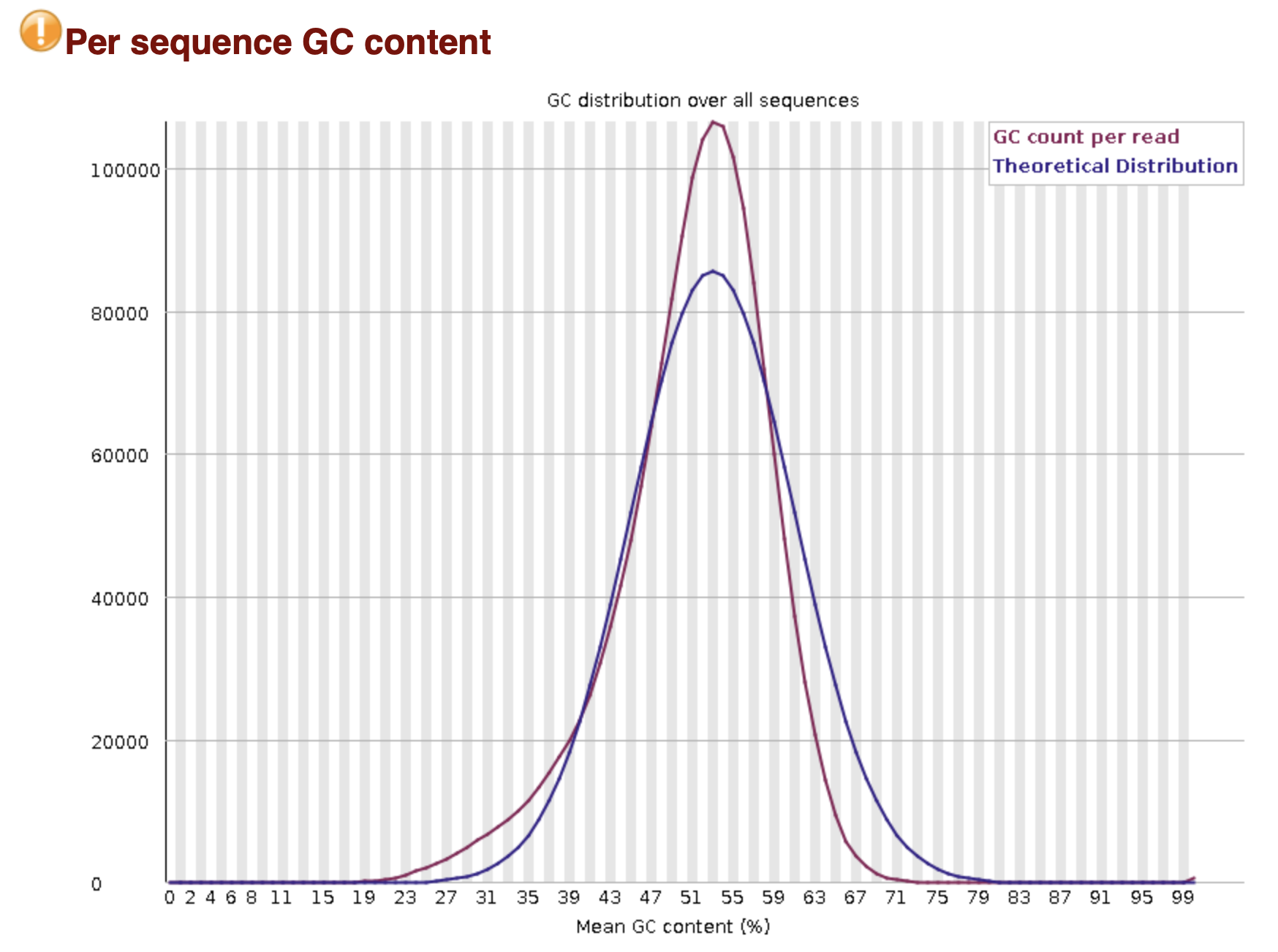
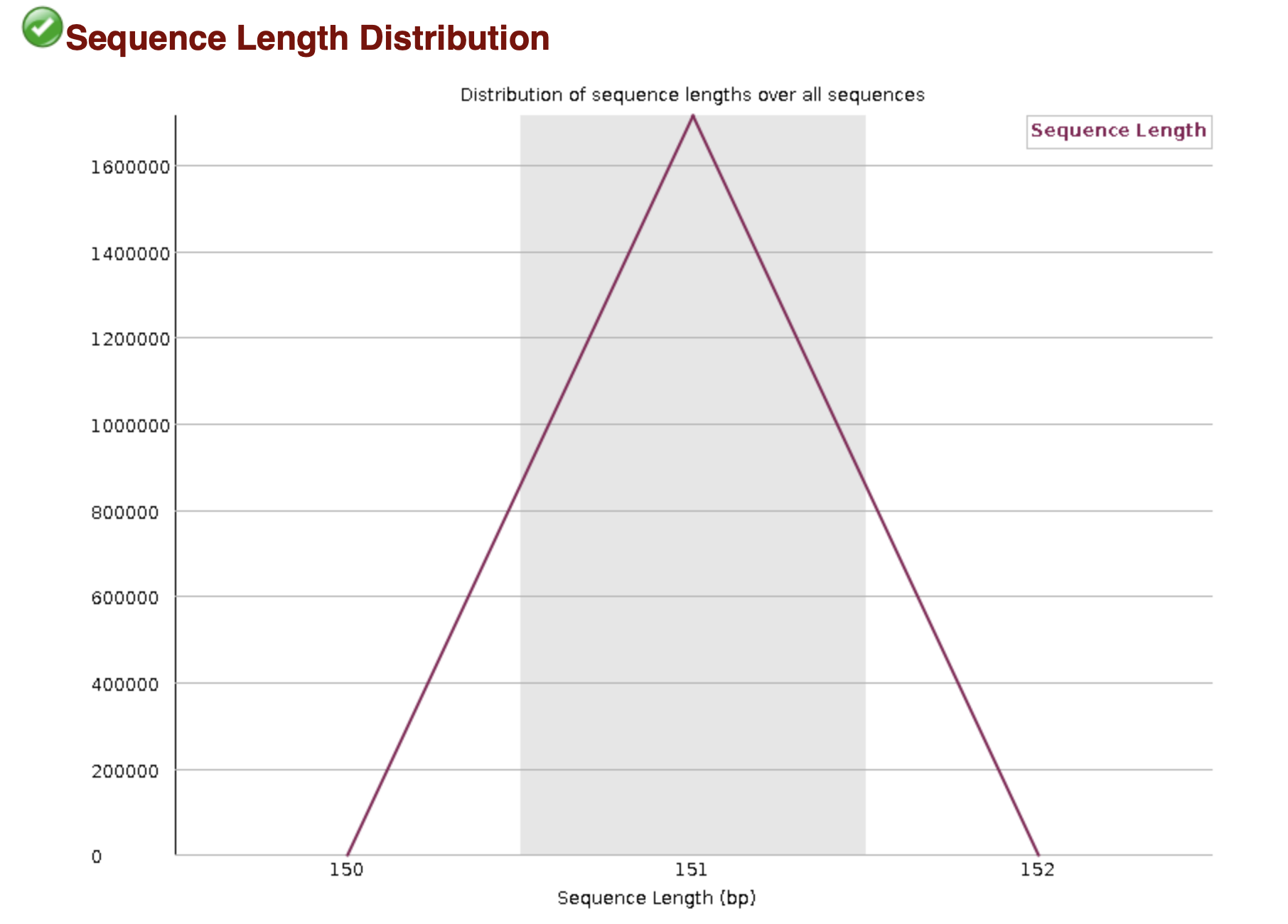
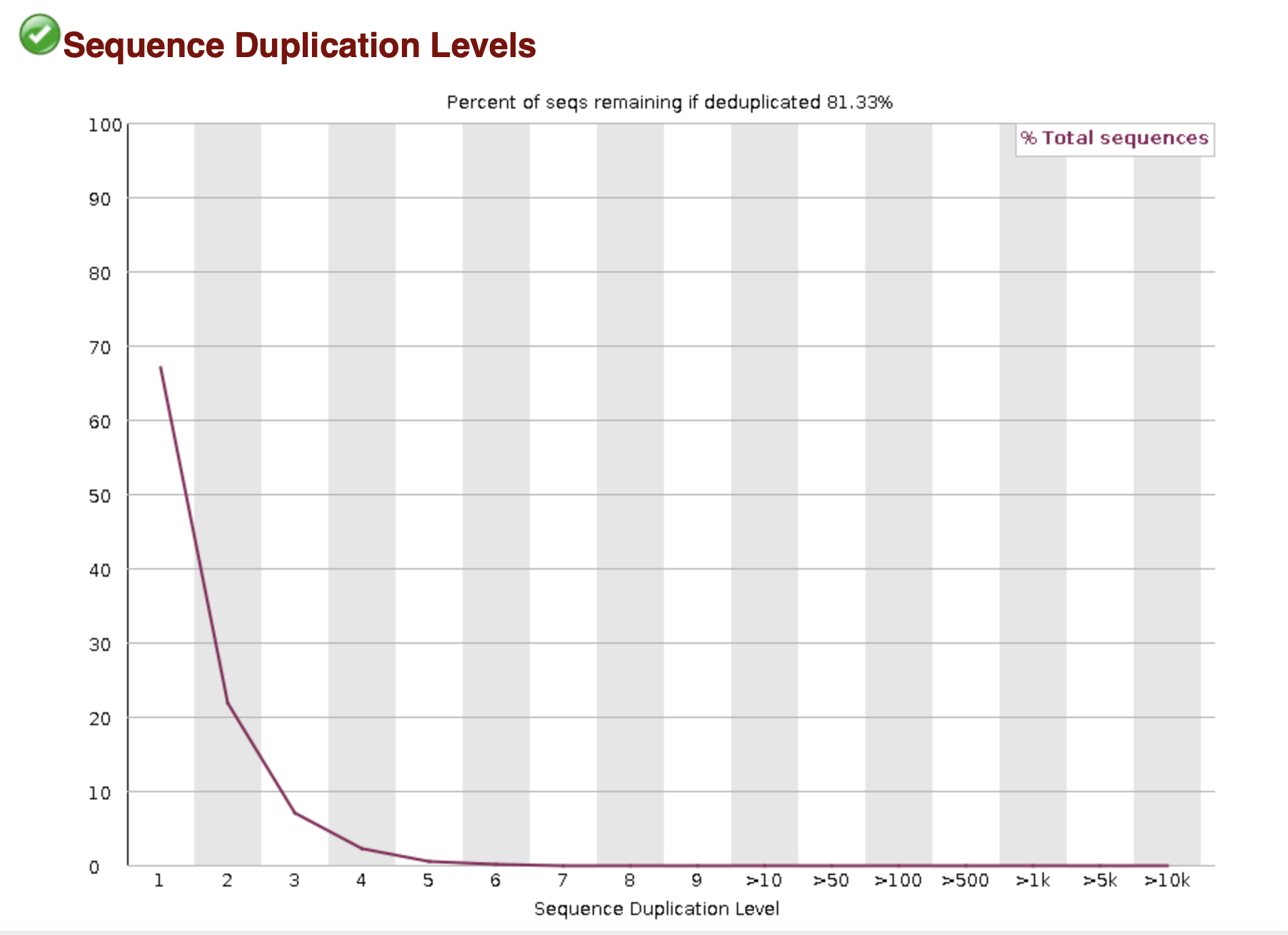
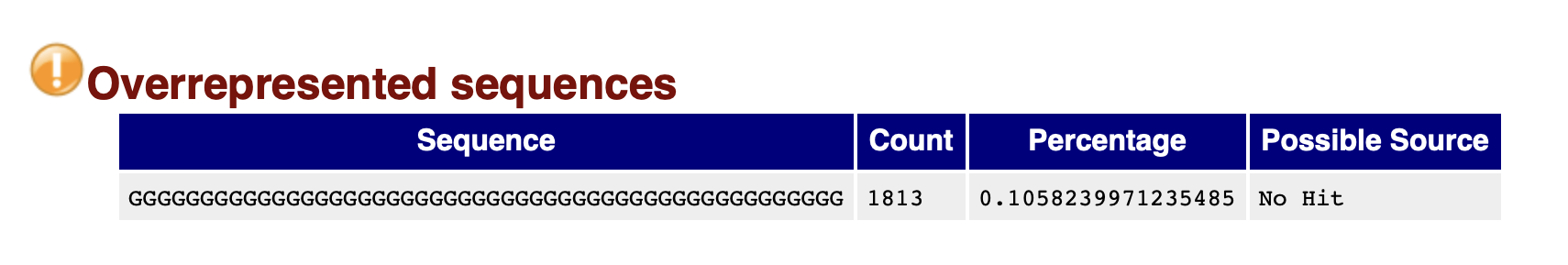
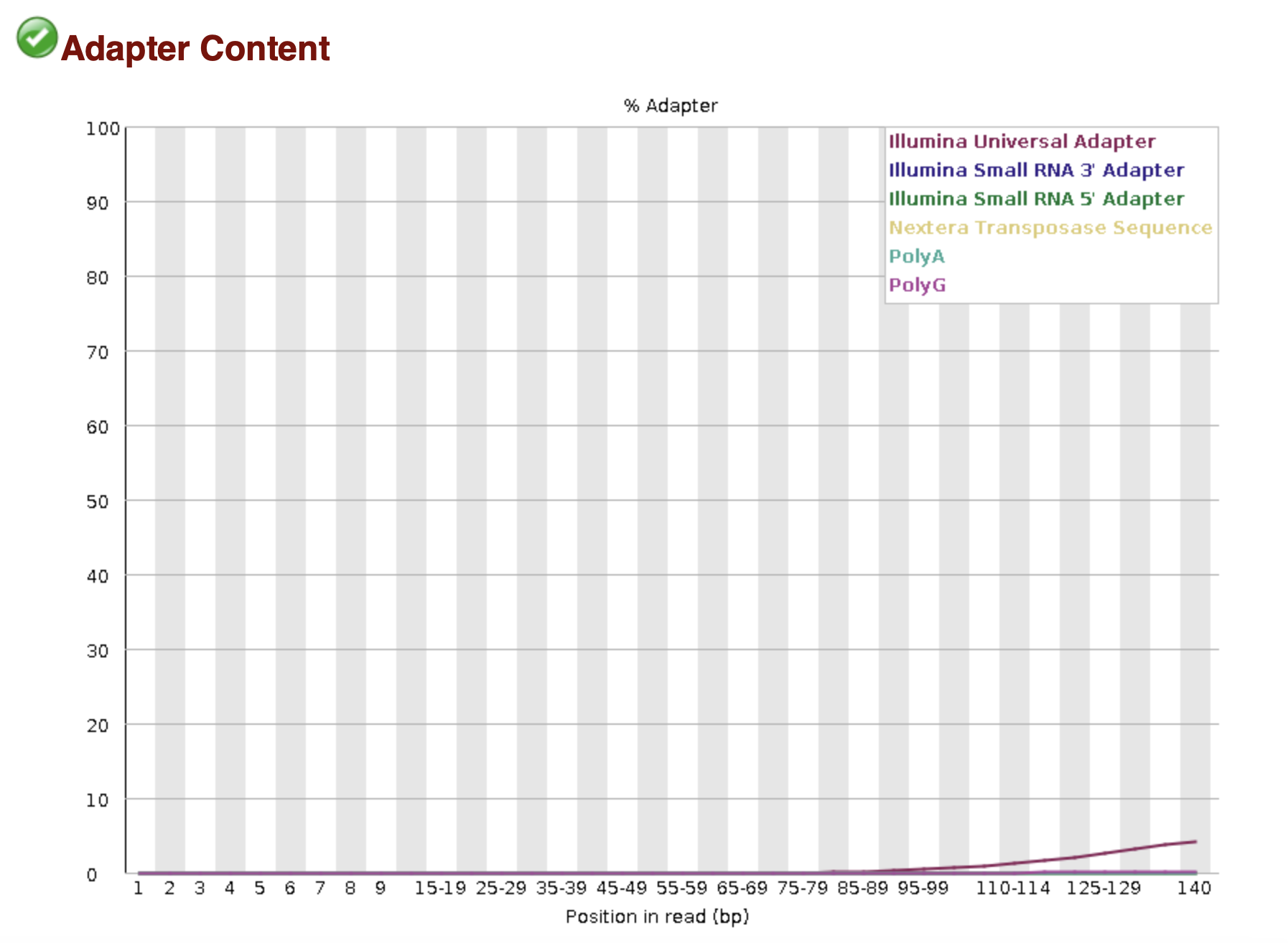
MultiQC
MultiQC compiles FastQC results together into one HTML file for easier comparison of samples.
To use MultiQC, the command needs a directory to output the results and the directory to search for the FastQC results to compile together:
multiqc -o multiqc_results raw_data/*fastqc.zipHomework Walkthrough
Log into your OSCER account
DIRECTIONS ON GETTING DATA
Next type the following command
nano qc_script.sh- Copy and paste the script below into your nano screen:
#!/bin/bash
#SBATCH --partition=ircfhp
#SBATCH --nodelist=c923
#SBATCH --container=el9hw
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=10
#SBATCH --mem=30G
#SBATCH --chdir=/home
#SBATCH --output=qc_%A_%a_stdout.txt
#SBATCH --error=qc_%A_%a_stderr.txt
#SBATCH --mail-user=USERo@ouhsc.edu
#SBATCH --mail-type=ALL
#==============================================================================
# BASH Strict mode (i.e. "fail fast" to reduce hard-to-find bugs)
set -e # EXIT the script if any command returns non-zero exit status.
set -E # Make ERR trapping work inside functions too.
set -u # Variables must be pre-defined before using them.
set -o pipefail # If a pipe fails, returns the error code for the failed pipe
# even if it isn't the last command in a series of pipes.
#==============================================================================
module load fastqc
module load multiqc
data_dir=""
fastqc_dir=""
multiqc_dir=""
mkdir $fastqc_dir
mkdir $multiqc_dir
fastqc -o fastqc_results raw_data/*.fastq.gz
multiqc -o multiqc_results raw_data/*fastqc.zipSubmission for Assignment
Run the following command to download the resulting MultiQC report to your local computer, submit this HTML file to Canvas:
scp USERNAME@dtn2.oscer.ou.edu:path/to/multiqc_report.html COMPUTER_DIR